-
Whitepaper
TAP® + Illumina NGS Platform enables High Quality Whole Exome Sequencing
HIGHLIGHTS
- The TAP is a non-invasive and virtually painless blood collection device that enables point-of-care, high-quality whole blood collections for downstream molecular applications such as human whole exome sequencing (WES).
- The Twist Biosciences Human Core WES kit enables cost-effective and comprehensive exome sequencing for germline (human diseases on Illumina sequencing instruments.
- Raw data concordance assessments for sequencing quality metrics such as target coverage, target coverage depth, aligned reads, uniformity, SNV variance, Q30 base quality (PHRED), DRAGEN enrichment metrics and other Picard metrics demonstrate quality control equivalence between TAP vs traditional venipuncture collections.
- Applications for efficient blood collection coupled to rapid WES enable remote access sequencing programs, germline disease research and diagnostics, genetic screening applications and more.

Figure 1: Overall workflow showing TAP blood collection through DRAGEN automated analysis.
INTRODUCTION
Human WES entails next-generation sequencing (NGS) of the entire protein-coding regions within the human genome. The human exome represents <2% of the entire genome while also containing ~85% of known disease-related variants. As a result, WES may sometime be a more cost-effective option and computationally data-efficient as compared to whole genome sequencing (WGS). While both are beneficial for comprehensive sequencing assessments, the cost of WES is rapidly decreasing with the introduction of innovative sequencing chemistries from novel probe-capture methods and the standardization of library preparation workflows via high-throughput automation. The Twist Bioscience Human Core WES kit enables WES of 33 Mb of CCDS coverage, including 99% of ClinVar variants and 99.3% of targeted regions covered at 20X with 5.3 Gb. By combining with an Illumina sequencing platform and downstream DRAGEN analysis, the possibilities for WES can include biomarker discovery, variant disease research, oncology applications, exonic genotype to phenotype assessments, population genetics and screening of other genetic diseases.
Traditional blood collection for WES involve a venipuncture draw by a trained phlebotomist, whereby 5-10 ml of whole blood is collected in a standard citrate/heparin/EDTA tube. This procedure can be relatively costly and inaccessible to scale across large patient populations, whereby economic and logistical hurdles to high-quality blood collections may persist due to the need of a trained phlebotomist. Here, we introduce the Yourbio Health TAP blood collection device as a viable tool to enable feasibility of blood collection scale and adoptability for downstream NGS on Illumina sequencing instruments. The TAP enables point-of-care collections by novice users, direct use by physicians/technologists in general practices, or collections in underserved/remote geographies. We obtained paired blood samples (TAP + traditional venipuncture) to analyze downstream Illumina NGS data to demonstrate analytical concordance for a variety of NGS quality metrics.
MATERIALS AND METHODS
Under an IRB-approved protocol, ten donors had paired blood samples collected by a trained phlebotomist (traditional venipuncture tube) and assisted-collections (TAP device) into heparin-coated tubes. The venipuncture tubes collected the full max fill volume of 5 ml by the phlebotomist, whereas the TAP devices collected ~500 ul of whole blood. All blood samples were collected and treated identically (same-day shipment to lab on ice packs during transit) in preparation for downstream DNA extraction and library preparation for WES. Briefly, 200 ul of whole blood was used as input volume for the ThermoFisher PureLink Genomic DNA Mini Kit, whereby 45 ul was used as the final elution volume. After extraction, DNA input was used per protocol instruction of the Twist Bioscience Human Core Exome WES Library Kit (see references). After library preparation, the final libraries were pooled in accordance with Azenta Life Science’s protocol and loaded onto an Illumina sequencer with the following parameters: CLIA Environment WES (performed by CLIA-trained personnel in a CLIA compliant lab using CLIA-qualified equipment) including sample QC, library CLIA-qualified equipment) including sample QC, library constructions and QC, Illumina sequencing to ~2.65Gb (estimated 30X) of data with FASTQ file delivery and raw data report, 2x150bp sequencing configuration, a quality guarantee of ≥80% of bases ≥Q30 scores.
Raw data analysis in an unbiased fashion was performed in the cloud (Illumina Base Space Sequence Hub – BSSH) by Dragen Enrichment v4.0.3 application. Briefly, the primary and secondary analysis was conducted in ~51 minutes using a multi-node computational session exercising a total of 20 nodes with a compute charge of 57 iCredits (~$57 total) with a total data delivery of 77.09Gb (FASTQ, BAM, VCFs generated for multiple callers). The germline small variant caller was used with the human reference genome (1000 Genomes HG38 ALT-masked V2). A custom BED equivalent to the Twist Bioscience Human Core Exome BED was used for Hg38 (see references for direct downloadable link). Base padding of 150 was used and QC coverage metrics were based on the Twist Bioscience BED file. The targeted region BED was also used as the probe BED. The following additional callers were enabled: multi-allelic filtering, HLA calling, SV calling, duplicate marking, Nirvana annotation. Lastly, the pipeline configuration was set to map/align + variant caller, whereby the small variant caller output included both the VCF and gVCF files.
RESULTS
DNA extraction QC assessments were conducted via two common methods: Nanodrop spectrometer and Qubit fluorescence quantification. Generally, A260/A280 ratios have ideal values ~1.6-2.1. All samples passed established CLIA-lab quality control checks. We show the Nanodrop CV of quality (A260/A280) in comparison to the CV for the total DNA amount (yield). Of note, traditional venipuncture tubes have higher total DNA yields (9/10 donors) than TAP samples. This may be due to possible differences in cellular homogeneity (capillary vs. venous cell populations), human operator handling of samples during extraction (input volumes for extraction), and the larger volumetric preservative containers for venipuncture. However, the average A260/A280 CV% across all samples is ~5% (range 0-11%), demonstrating that the DNA quality between the two sample collection modalities are comparable. Overall, Qubit concentrations were used for all downstream library preparation and NGS calculations. (Note: RNA Qubit concentrations are randomly assessed via cherry picking of samples (n=5 per batch), whereby RNA contamination percentage = RNA Qubit yield / DNA Qubit yield. This crude assessment is a random metric used to monitor extraction quality from batch to batch. One sample (003-Heparin) showed 9.21% RNA contamination – this does not affect the downstream performance for germline DNA WES.)
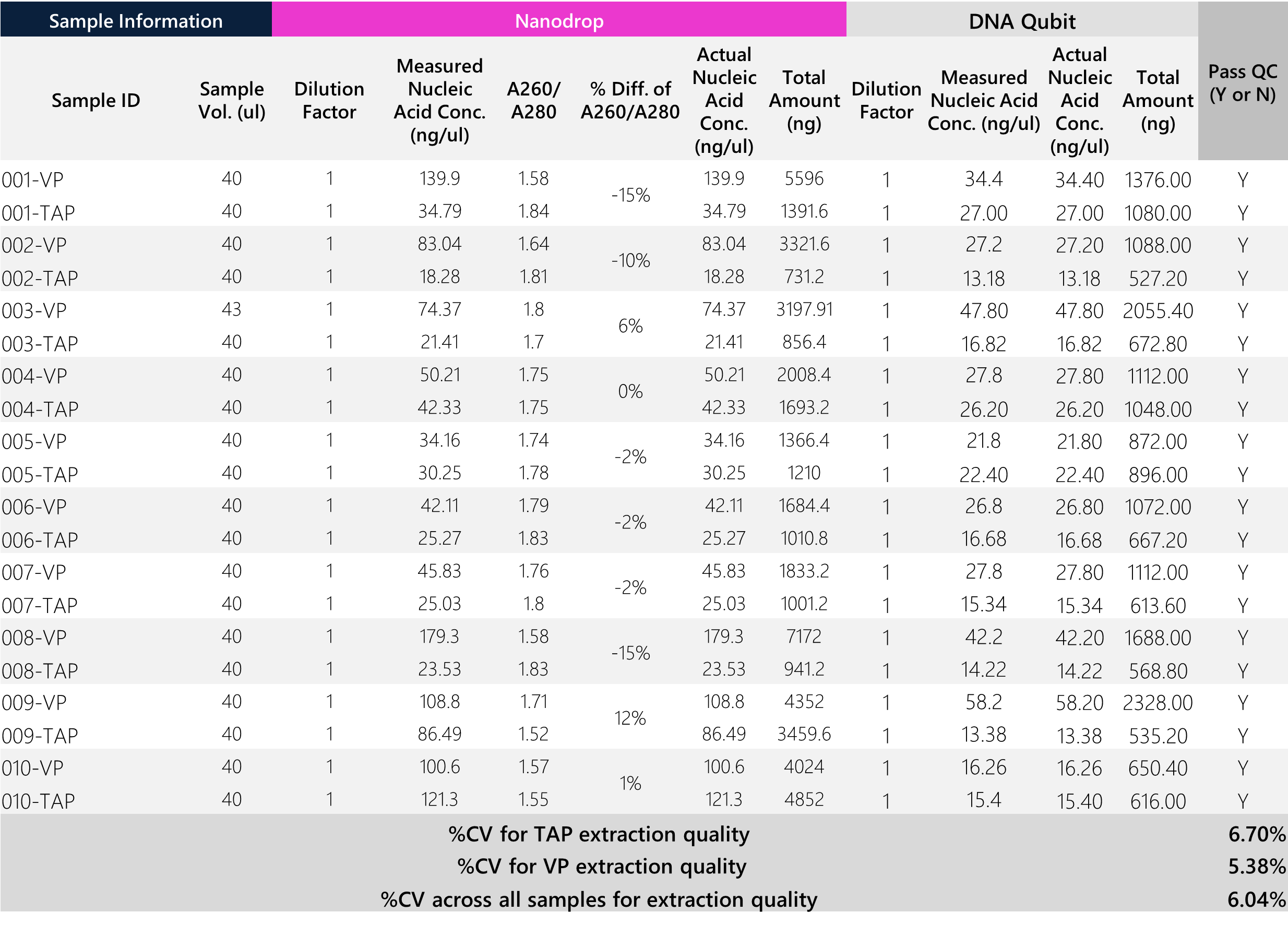
Table 1: Tabular data for DNA sample QC via Nanodrop and Qubit assessments. All samples passed CLIA-lab QC.
Raw data analysis of sequencing was performed by Illumina Dragen Enrichment v4.0.3 pipeline within BSSH, whereby read level, base level, alignment/coverage, variant/SNVs detection, base-quality pass-rates, uniformity of coverage (percent > 0.2 of mean), coverage depth, target coverage at 1X-20X (here, WES was performed with a sequencing guarantee of 30X), fragment length median, read duplication percentage and more were compared between both sample types (TAP vs. traditional venipuncture). Complete raw data excel reports for Dragen Enrichment summaries and the sequencing Picard metrics are available as supplementary files upon request. Select values are presented below and categorized by read level enrichment, base level enrichment, coverage, fragment quality, variant caller, and bait summaries.
Read level metrics between TAP and VP show enrichment efficiencies of <10% differences across paired samples). The average total aligned reads, unique aligned reads and target unique aligned reads also demonstrate <7% differences between TAP and VP. Overall, the quality of read enrichment is comparable between both methods.
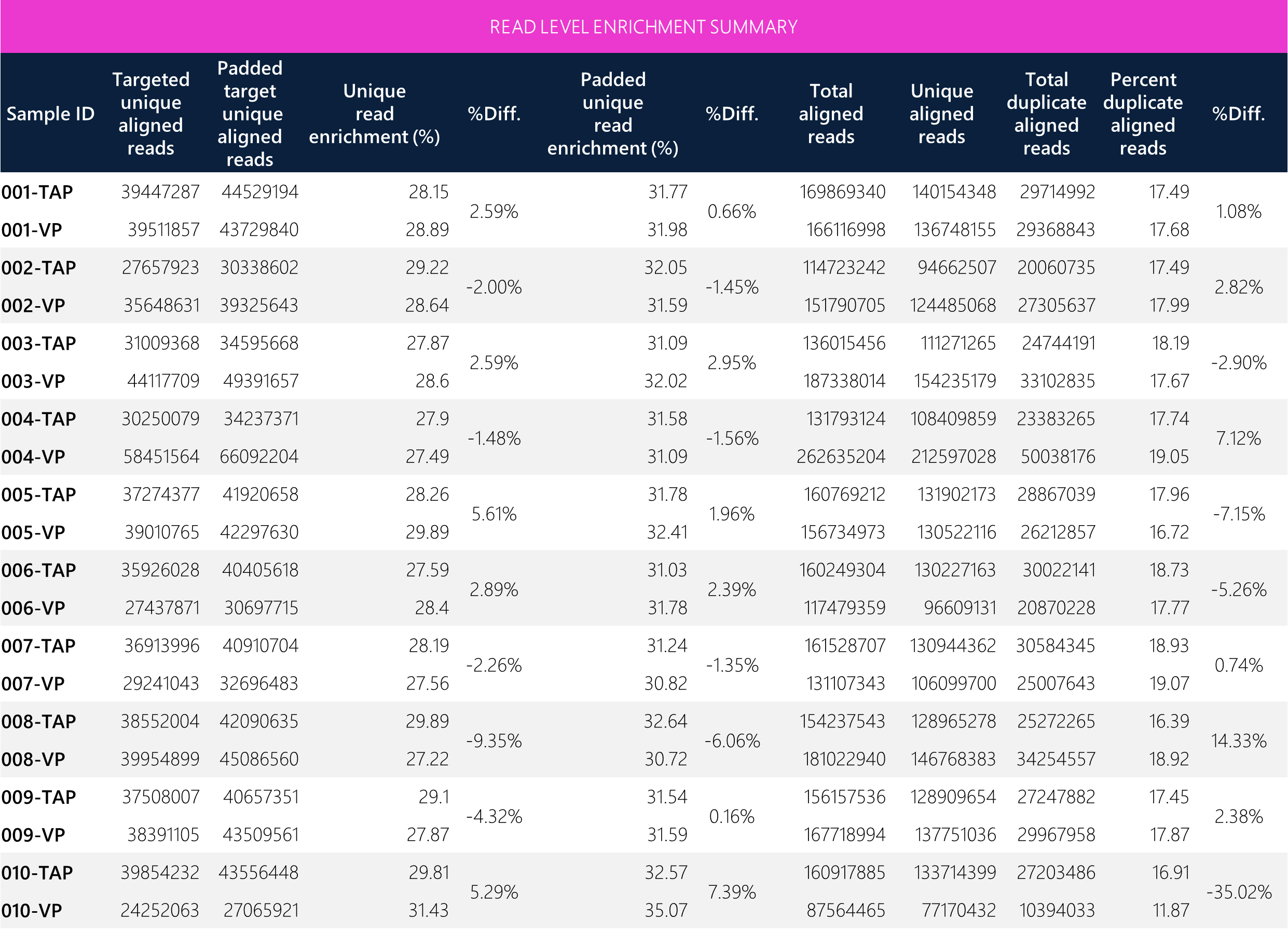
Table 2: Tabular data for read level enrichment between paired TAP and VP samples.

Table 3: Summary of means and overall average percent difference between TAP and VP samples.
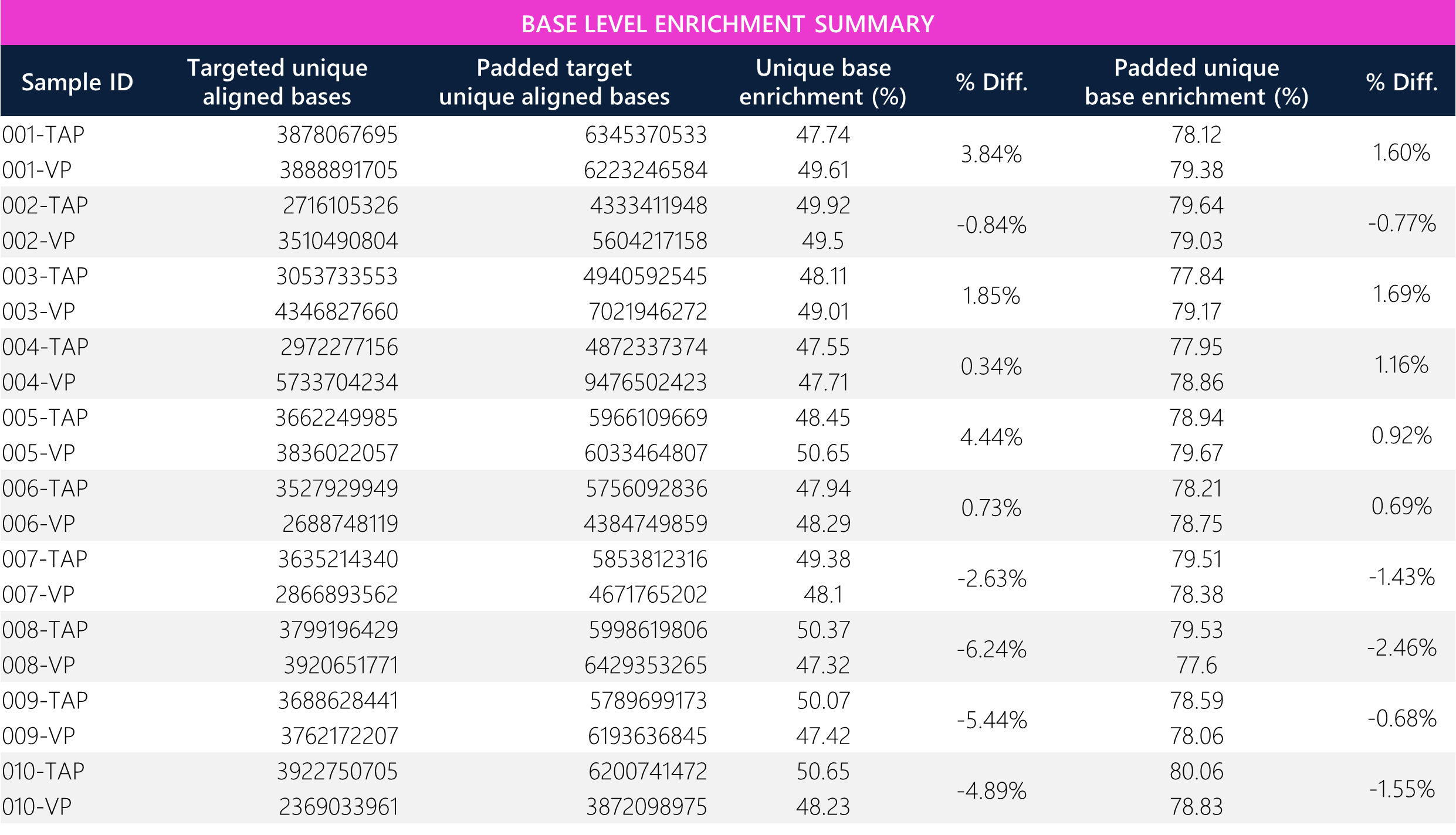
Table 4: Tabular data for base level enrichment between paired TAP and VP samples.
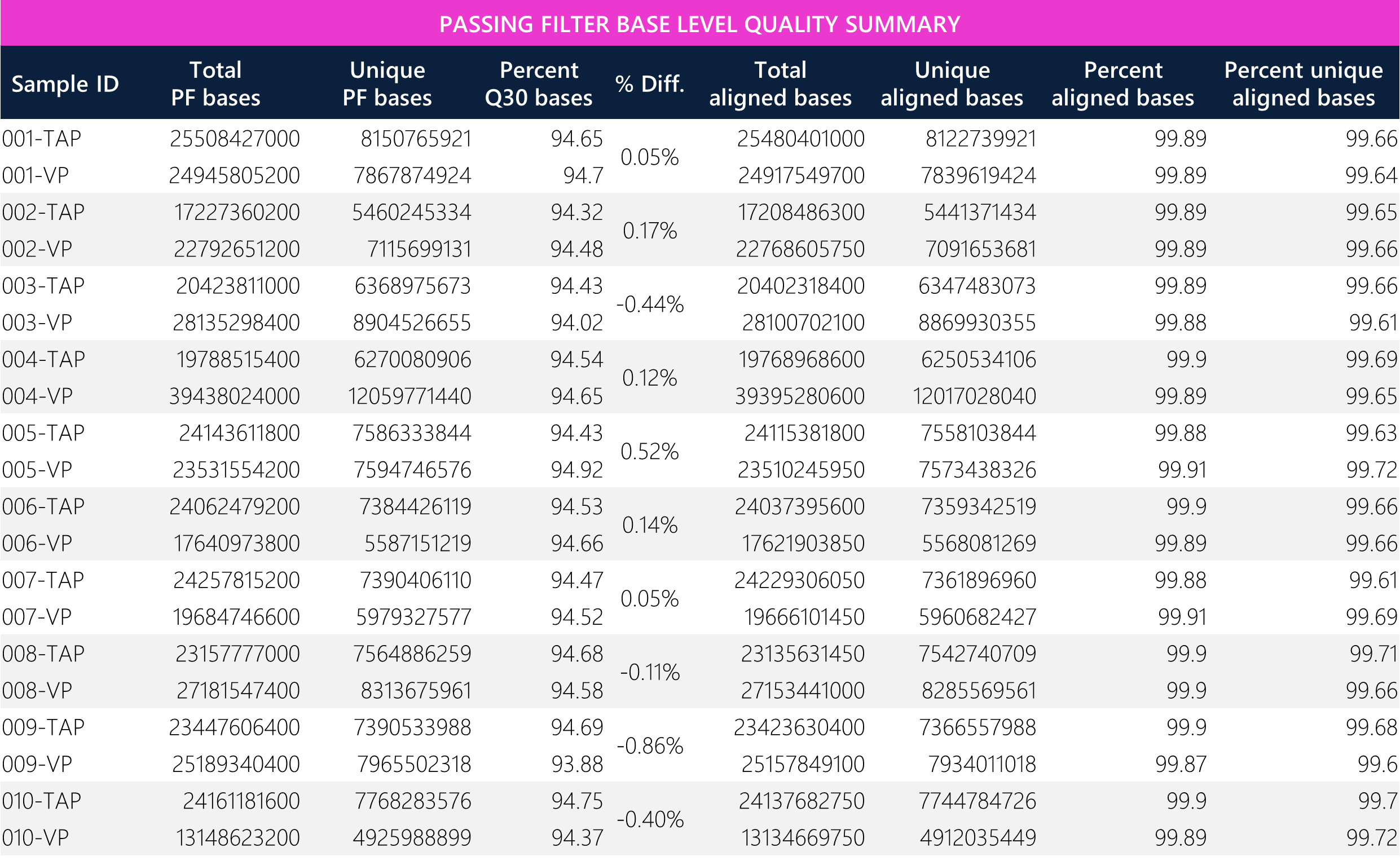
Table 5: Tabular data for passing filter base level quality metrics between paired TAP and VP samples.
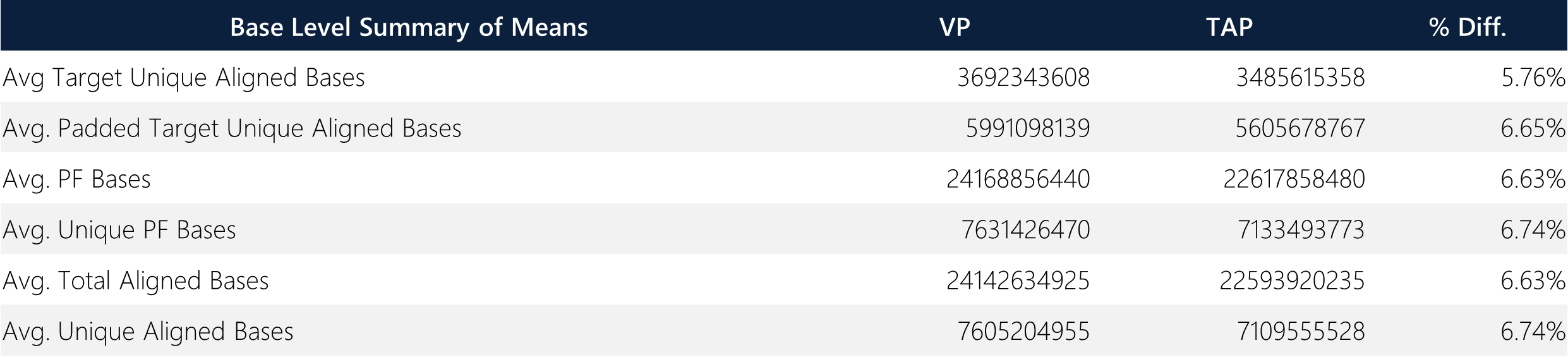
Table 6: Summary of means and overall average percent difference between TAP and VP samples.
Padded unique base enrichment efficiency shows <3% differences across all paired samples. Notably, the passing filter bases, which are defined as Q30 bases, show <1% differences across all paired samples (acceptance criteria and sequencing guarantee = >80%; this run ~94%). All samples demonstrate >99.5% unique base alignment. Overall, the average target/padded unique aligned bases, unique/passing filter bases, and total/unique aligned bases show <7% differences between TAP and VP methods, demonstrating comparability between both sample types.
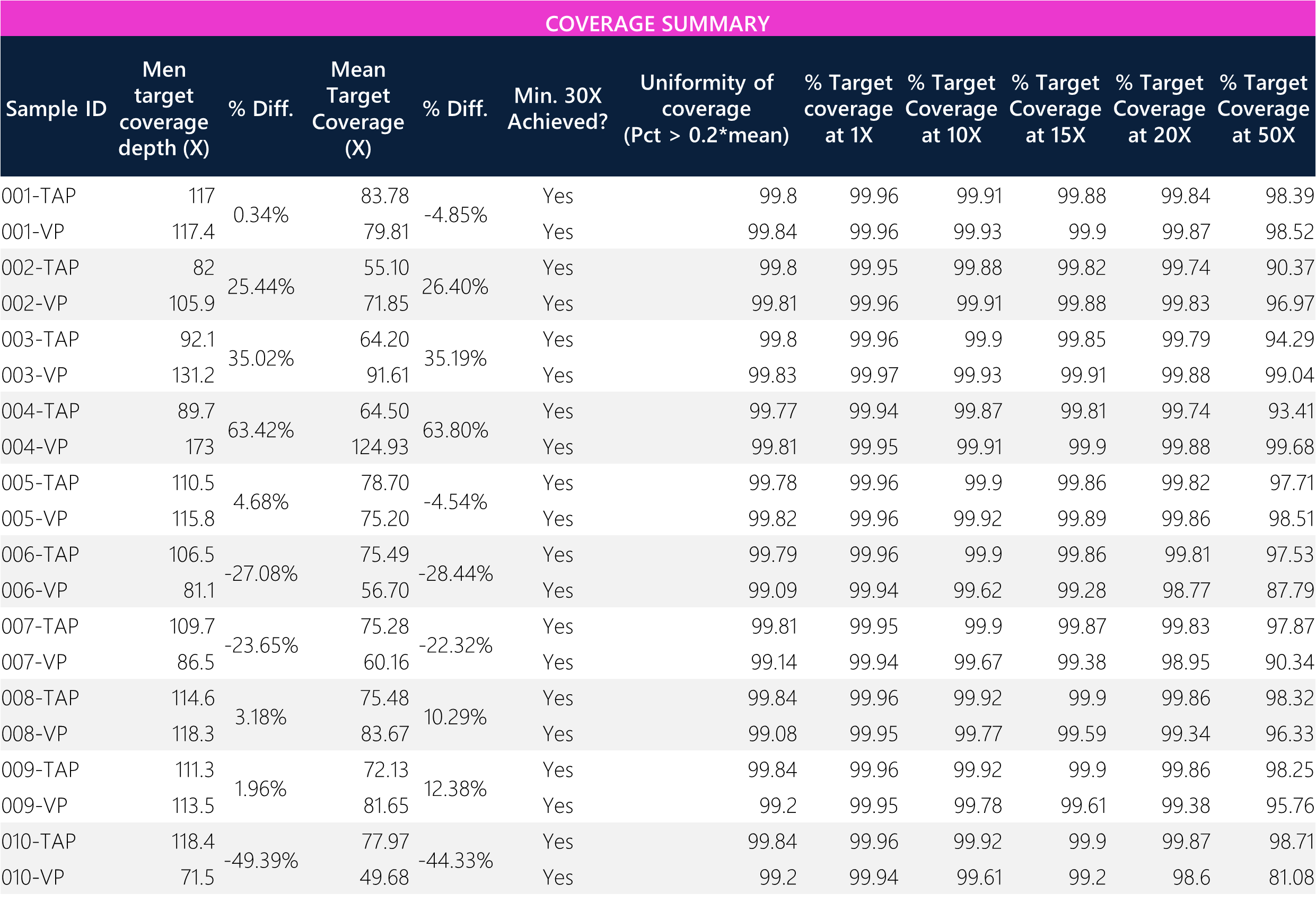
Table 7: Tabular data for coverage metrics between paired TAP and VP samples.

Table 8: Summary of means and overall average percent difference between TAP and VP samples.
Coverage differences between paired samples show high variance. This sequencing run had an acceptance criteria (sequencing guarantee) of 30X target coverage for WES, whereby all samples achieved the minimum threshold. Overall, the average target coverage is approximately >2x the sequencing guarantee, with ~7% difference between VP and TAP. Additionally, there is ~99% target coverage at 20X, with a slight reduction at 50X for some samples (in line with the 30X sequencing guarantee). The uniformity of coverage across all samples is >99%, indicative of good read depth quality between both collection methods.
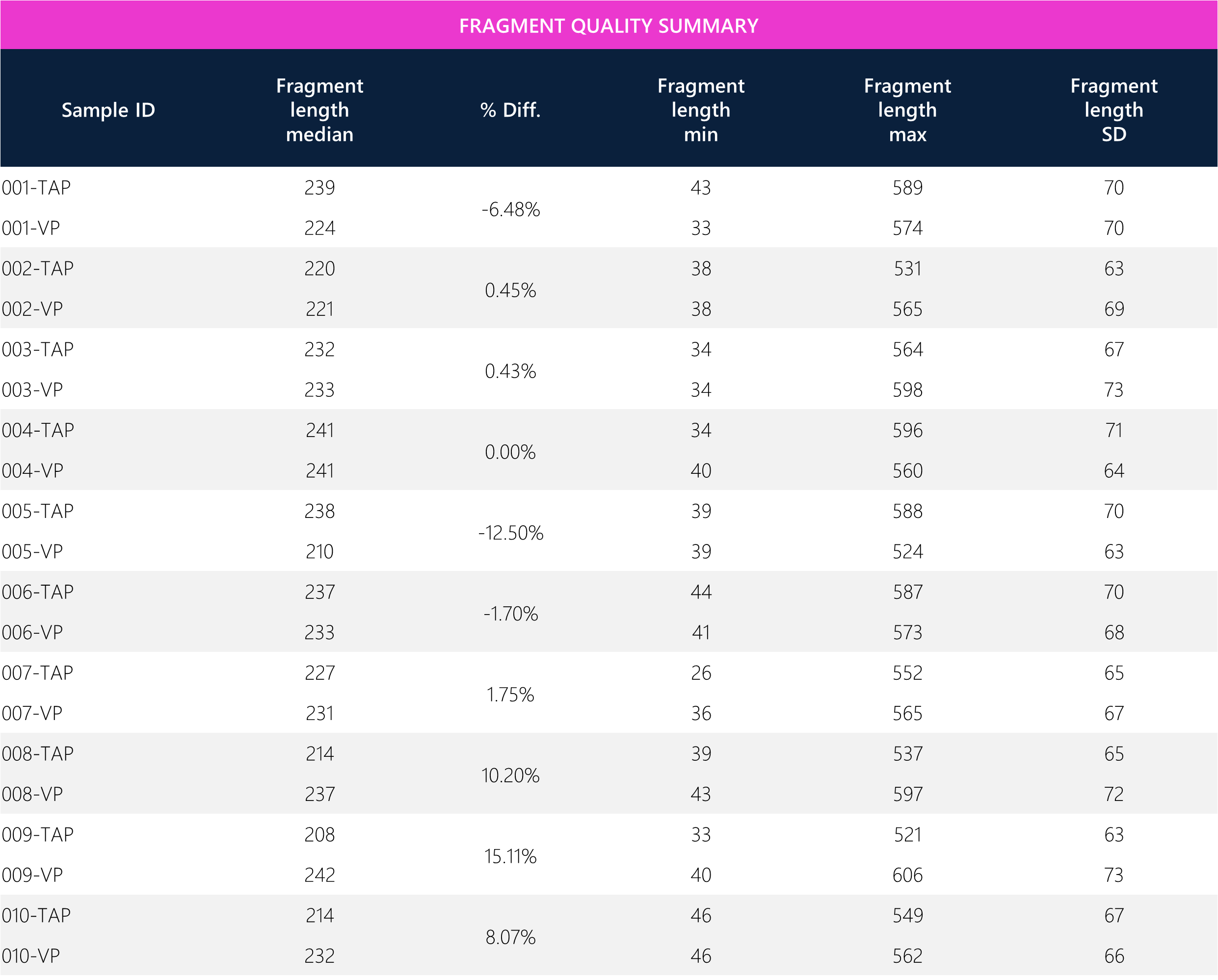
Table 9: Tabular data for fragment quality between paired TAP and VP samples.

Table 10: Summary of means and overall average percent difference between TAP and VP samples. The overall average fragment length median between VP and TAP is <2%, whereby 2×150 bp cycle configuration was used (300bp max length per read).
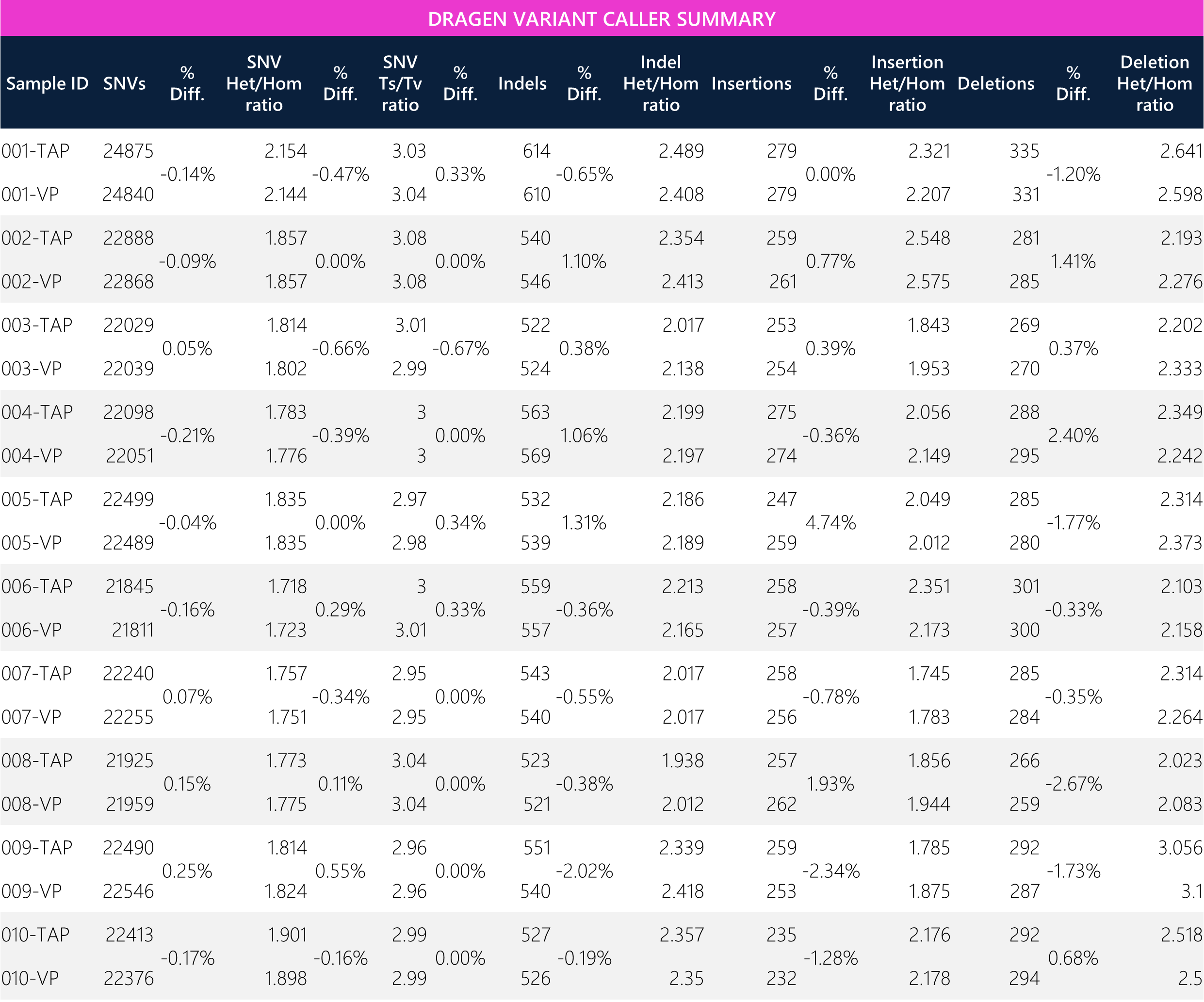
Table 11: Tabular data for variant classes between paired TAP and VP samples.

Table 12: Summary of means and overall average percent difference between TAP and VP samples.
Secondary analysis was conducted via Dragen bioinformatics pipeline. Raw data analysis was primarily intended for single nucleotide variants (SNVs) while inclusive of insertions/deletions (indels), insertions and deletions for additional value. The aim is to show potential differences in SNVs and other variant classes from automated classification (without human/tertiary analysis) to demonstrate comparability of calls. Paired samples show less than ~0.25% differences for SNVs, while the average differences across SNVs between collection matrices is ~0.03%. Overall, the average percent differences across all variant classes between TAP and VP is approximately <0.31%, demonstrating quantitative comparability of calls between matrices. (Note: VCF comparisons between paired samples are discussed elsewhere.)
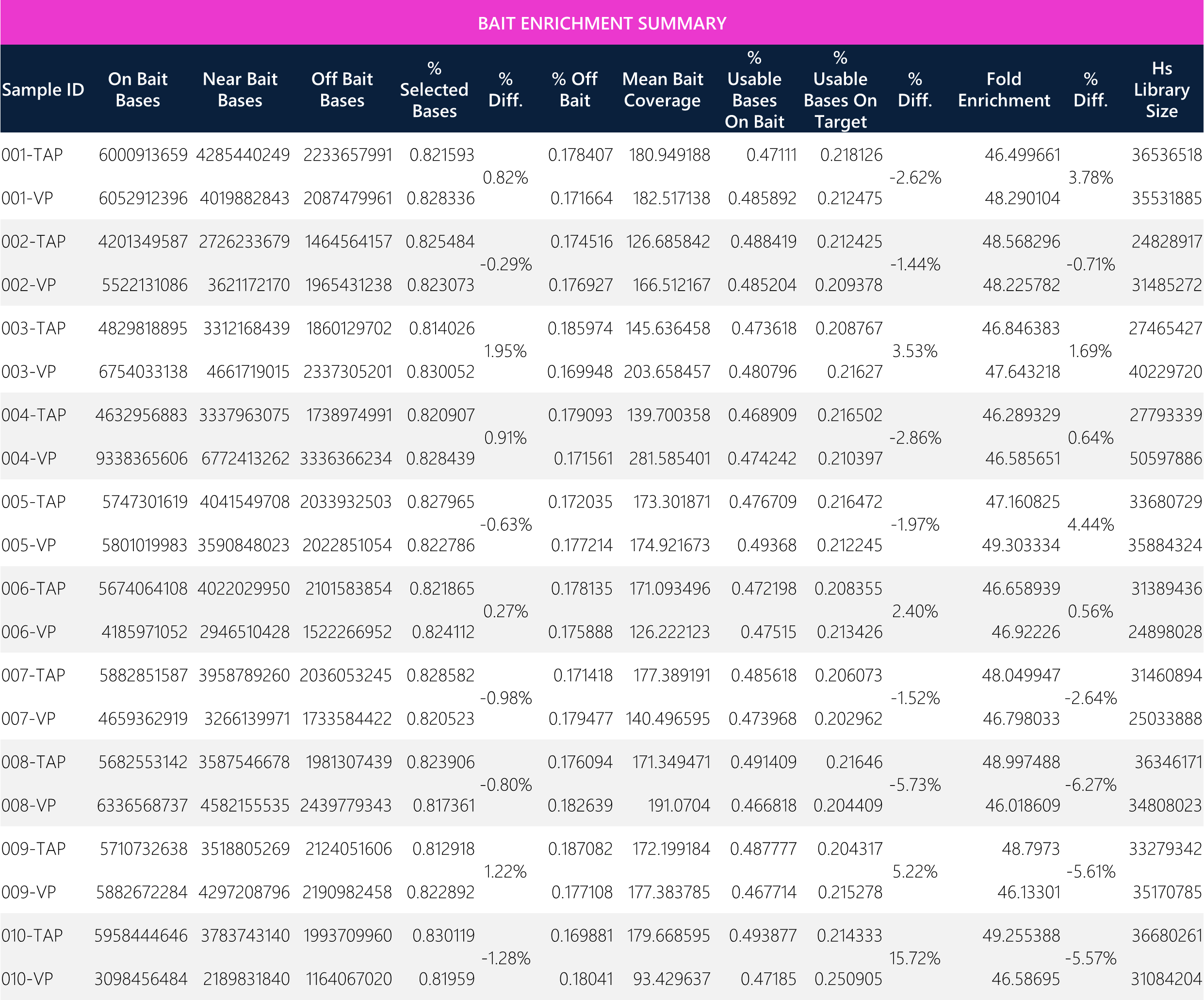
Table 13: Tabular data for bait enrichment between paired TAP and VP samples.

Table 14: Summary of means and overall average percent difference between TAP and VP samples.
The Twist Exome Core (hg38) bait set was used for the wet lab library preparation with a total bait territory consisting of 33,163,529 bases (~33.2 MB). WES analysis was conducted on a human genome reference size of 3,217,346,917 bases. Importantly, the average mean bait coverage shows <6% difference between sample matrices.
CONCLUSIONS
Initial feasibility for WES using Illumina-based NGS and the Twist Bioscience library kit demonstrate good concordance of sequencing quality assessments between TAP and VP samples. Specifically, Picard and Dragen enrichment metrics highlight multiple quality measures at the sample extraction, library preparation, and sequencing stages (primary and secondary analyses). Accessible and minimally invasive sampling with the TAP device can enable comprehensive genetic testing over more frequent timepoints with rapid access to familial genetic trees. Remote collections with TAP can enable access, throughput and automated data analysis when coupled to sensitive and specific platform technologies such as NGS via Illumina instruments and cloud-based genomic pipelines. The purpose of this study is to demonstrate technology and platform capability of the TAP device with the Illumina platform using the Twist Bioscience Human Core Exome library kit. Further evaluation of the TAP device could benefit from a larger sample size and comparisons to other sequencing technologies and sample collection methods such as dried blood spots (DBS) and saliva. Future studies will explore other collection mediums such as saliva collection devices and DBS cards in addition to whole genome sequencing and low-pass sequencing. Additional consideration may be inclusion of cellular heterogeneity assessments between TAP and VP samples for germline and somatic assessments.