-
Whitepaper
Enable remote access multiomics with just one TAP®
Olink Proteomics
HIGHLIGHTS
- The TAP is a minimally invasive and virtually painless blood collection device that enables point-of-care, efficient whole blood collections for downstream proteomics applications such as multiplex immunoassays.
- The Olink Target 96 Cardiovascular II Panel enables targeted multiplexed detection of 92 specific proteins related to cardiovascular disease (CVD) from as little as 1 ul of plasma/serum by implementing proximity extension assay (PEA) technology.
- Relative data concordance assessments for protein measurement quality and abundance was primarily assessed via the NPX (normalized protein expression) metric, linearization of NPX, QC metrics (average %CV, intra-assay %CV distribution, expected detectability, technical pass rate and QC warning rate) and pairwise Pearson/Spearman correlations between TAP vs traditional venipuncture collections.
- Applications for efficient blood collection coupled to comprehensive proteomics enables remote access sequencing programs, germline disease research and diagnostics, genetic screening applications and more.
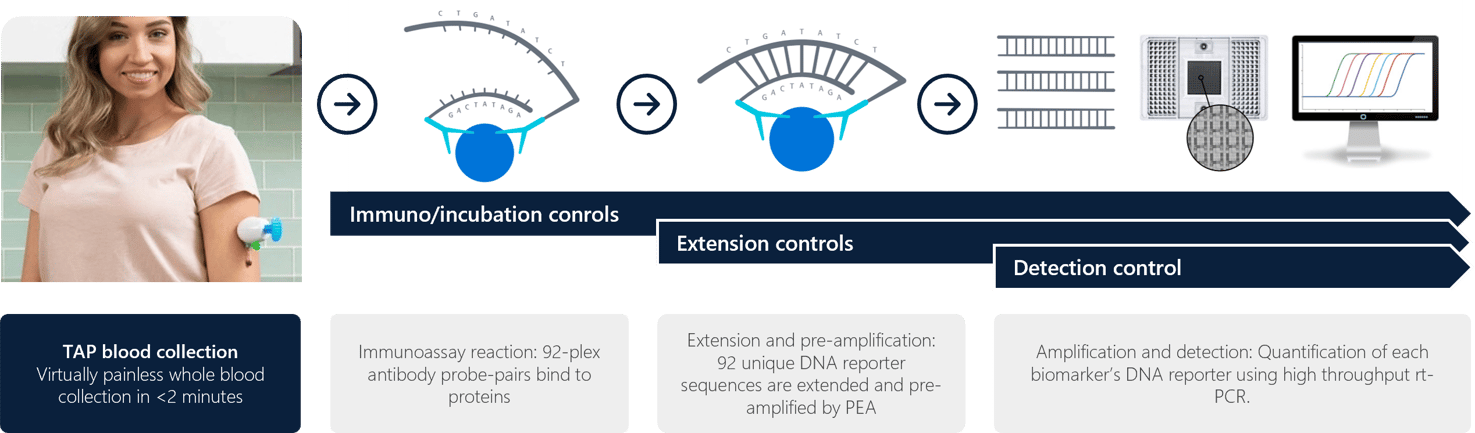
Figure 1: Overall workflow showing TAP blood collection to Olink PEA extension and qPCR readout.
INTRODUCTION
As genomics assessments continue to reduce in cost, the respective proteomics assessments are emerging as the next level of understanding complex real-time human biology. Biomarker discovery for proteins enables identification of protein signatures related to pathophysiological significance. This enables correlation assessments between genotypes and phenotypes in a comprehensive fashion. Technologies that enable broad yet specific, reliable, high throughput, precise and sensitive assessments from microsample volumes with low costs per datapoint enable both diagnostic and monitoring applications for precision medicine and routine healthcare.
Multiplexed and targeted protein expression on small sample volumes enables relative quantification for real-time monitoring and timepoint interrogations for relative changes in biomarkers of interest. By using only 1 ul of serum or plasma, the Olink Target 96 CVD II panel can assess 92 specific analytes simultaneously (plus multiple controls) by performing sample reactions in a standard 96-well plate with subsequent microfluidic qPCR on a Fluidigm Biomark instrument for high throughput assessments.
The Olink Target 96 panel and reagent workflow is foundationally based on PEA technology, where 92 oligonucleotide labeled antibody probe pairs are allowed to bind to their respective target proteins (if present within sample). A proximity-dependent DNA polymerization event enables the formation of PCR reporter sequence. This reporter sequence is amplified via thermocycling, whereby detection and quantification is accomplished via real-time PCR. All steps are standardized and efficiently conducted within a 96-well format. Data analysis is predicated on the Olink arbitrary defined metric of Normalized Protein eXpression (NPX) values, where statistical and relative quantification assessments can be calculated.
Traditional blood collection for massively parallel proteomics involve a venipuncture draw by a trained phlebotomist, whereby 5-10 ml of whole blood is collected in a standard citrate/heparin/EDTA tube. This procedure can be relatively costly and inaccessible to scale across large patient populations, whereby economic and logistical hurdles to high-quality blood collections may persist due to the need of a trained phlebotomist. Here, we introduce the Yourbio Health TAP blood collection device as a viable tool to enable feasibility of blood collection scale and adoptability for downstream Olink proteomics investigations. The TAP enables point-of-care collections by novice users, direct use by physicians/technologists in general practices, or collections in underserved/remote geographies. We obtained paired blood samples (TAP + traditional venipuncture) to analyze downstream Olink Target 96 data to demonstrate analytical concordance for a variety of Olink quality metrics.
MATERIALS AND METHODS
Under an IRB-approved protocol, ten donors had paired blood samples collected by a trained phlebotomist (traditional venipuncture tube) and assisted-collections (TAP device) into heparin-coated tubes. The venipuncture tubes collected the full max fill volume of 5 ml by the phlebotomist, whereas the TAP devices collected ~500 ul of whole blood. All blood samples were collected and treated identically (same-day shipment to lab on ice packs during transit) in preparation for downstream proteomics. Briefly, all whole blood collections were centrifuged to obtain plasma, whereby 1 ul of the resulting plasma was used as input volume for the Olink Target 96 CVD II panel (Table 1). Wet-lab setup was performed according to Azenta Life Science’s and Olink’s SOP in a CLIA environment performed by CLIA-trained personnel in a CLIA compliant lab using CLIA-qualified equipment (short instructions found here: https://olink.com/content/uploads/2022/05/olink-target-96-short-instructions.pdf):
- Incubation: a standardized incubation mix was prepared for a 96-well plate followed by transfer of 1 ul of sample per well, 1 ul of negative controls (triplicate), 1 ul of interplate control (triplicate), and 1 ul of pooled plasma sample (duplicate). An overnight incubation at 4C was performed.
- Extension: a standardized extension mix was prepared to transfer 96 ul of the extension mix to the incubation plate followed by thermocycling in a PCR machine via the PEA program.
- Detection: a standardized detection mix was prepared to transfer into a sample plate, whereby the incubation plate contents were transferred to the sample plate followed by transfer of primer plate and sample plate into the primed inlets of the Fluidigm Biomark (Olink Signature Q100 instrument) to run analysis.
Data analysis can be performed via softwares such as Olink Insight, Olink Power Tool, Olink NPX Signature, Olink Statistical Analysis app, Olink Analyze and other free and customizable tools (see here: https://olink.com/products-services/data-analysis-products/). The analysis in this paper was conducted from the standard NPX readout obtained from NPX Signature, whereby input data from the Signature Q100, Ct values exported from the Fluidigm Real-Time PCR Analysis software, NPX Manager .oaf project files and NPX Signature .npx study files were incorporated. After quality control and normalization against the Extension Control, the Inter-Plate Control (IPC) and a correction factor, the output data were obtained in Normalized Protein eXpression (NPX) values. NPX is an arbitrary unit on a log2 scale, where a high NPX value corresponds to high protein concentration. The NPX data file (excel) was used for all statistical analysis in this study (Table 2).
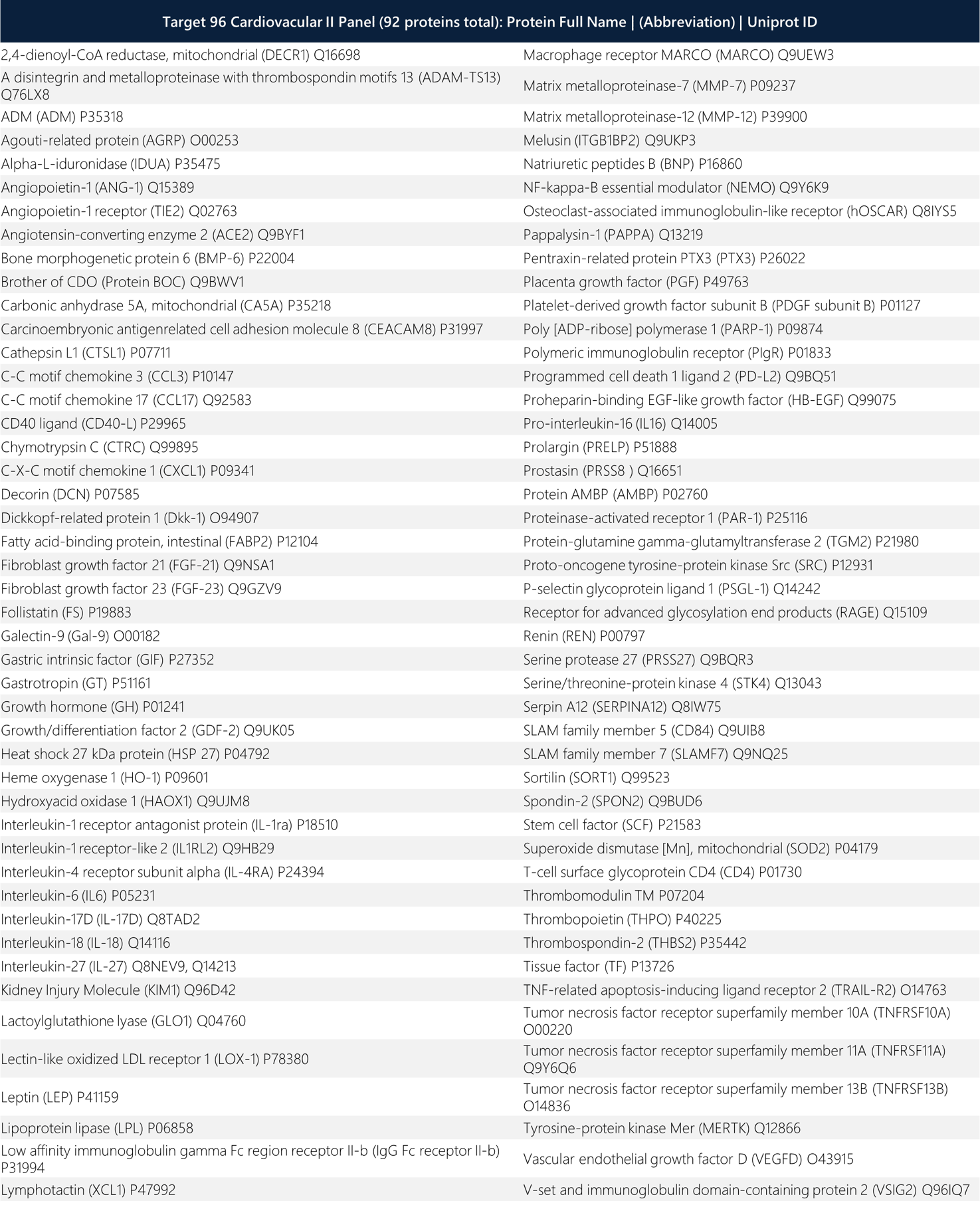
Table 1: Full names of protein biomarkers with Uniprot ID in the Olink Target 96 CVD II Panel.
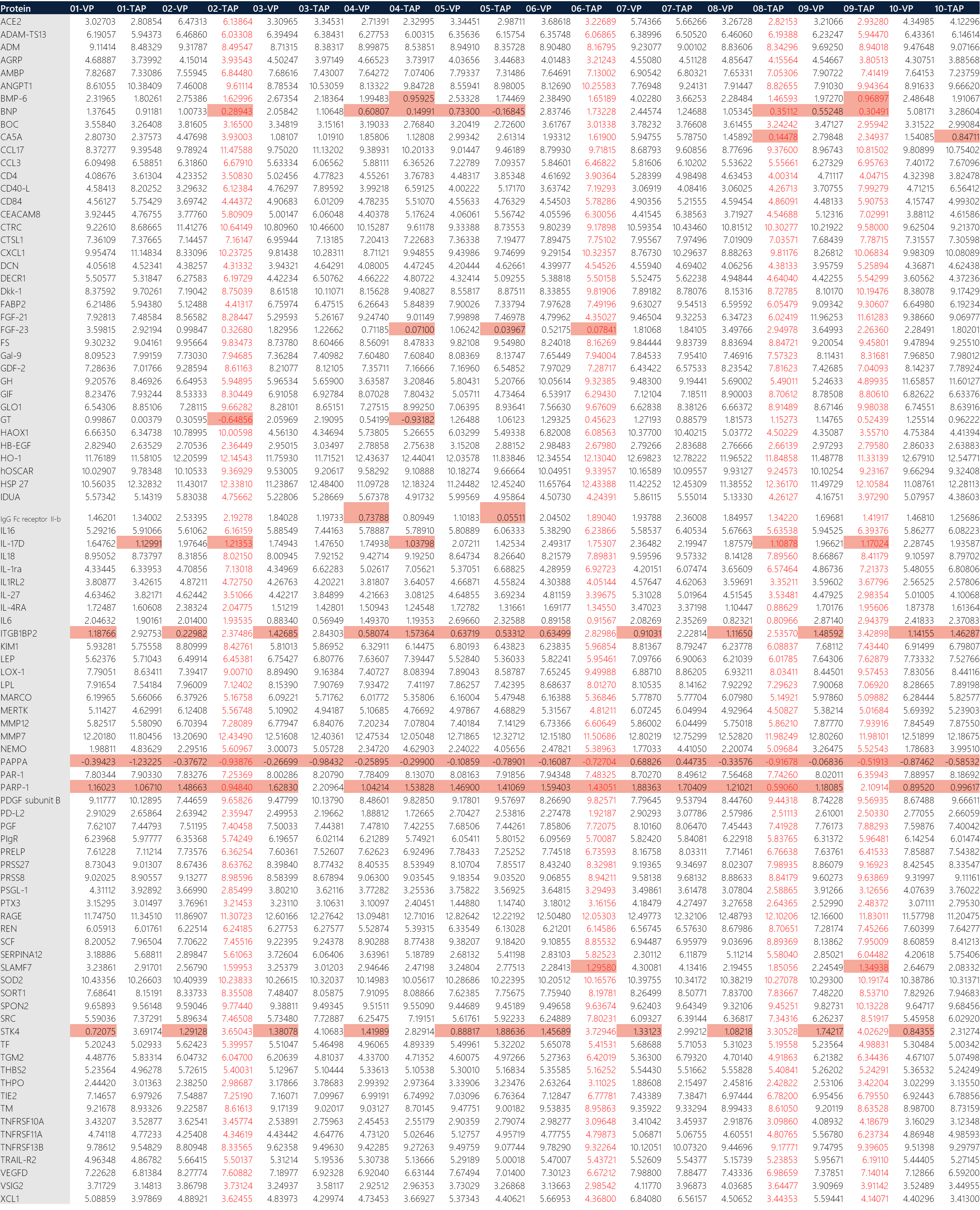
Table 2: Transposed raw data of NPX output from Olink Target 96 CVD II run. (Red text: samples that did not pass QC. Red fill: values below LOD for the respective protein biomarker.)
RESULTS
Briefly, microfluidic PCR of the Olink Target 96 panel generated Ct values for amplified products of PEA. The Ct values from qPCR were translated into the relative quantification unit known as Normalized Protein eXpression (NPX) values via a series of mathematical normalization calculations. Interpretation of data is based on the generated NPX for a particular panel as shown in Table 2. Data points for samples that did not pass QC are in red text, whereas cells containing data values below LOD are indicated with a red background. Data may still be used for samples with QC warnings and were treated with caution. The QC is a 2-step process:
- Each sample plate is evaluated on the standard deviation of the internal controls. This should be below 0.2 NPX. Only data from sample plate that pass this quality control will be reported.
- The quality of each sample is assessed by evaluating the deviation from the median value of the controls for each individual sample. Samples that deviate less than 0.3 NPX from the median pass the quality control.
A box plot and an inter-quartile range (IQR) plot for all NPX data was generated to show distribution of values, ranges and standard deviation for all samples. As expected, samples have a wide IQR due to the uniform assessment of all proteins.
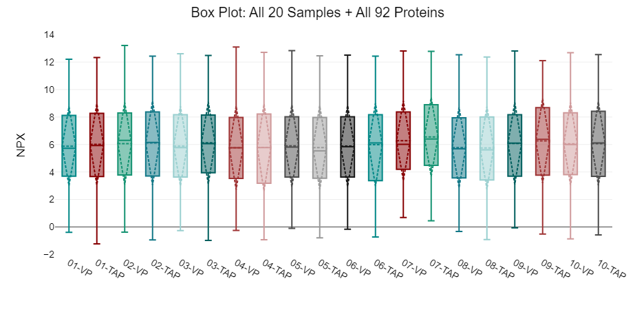 Figure 2: Box plot showing general distribution of all 92 NPX values across all 20 samples. Data is displayed as a box and whisker plot. Lower and upper ends of the boxes represent the 1st and 3rd quartiles of the data. The center line represents the median. Lower and upper fences show the minimum or maximum observed value that is within 1.5 times the inter-quartile range.
Figure 2: Box plot showing general distribution of all 92 NPX values across all 20 samples. Data is displayed as a box and whisker plot. Lower and upper ends of the boxes represent the 1st and 3rd quartiles of the data. The center line represents the median. Lower and upper fences show the minimum or maximum observed value that is within 1.5 times the inter-quartile range.
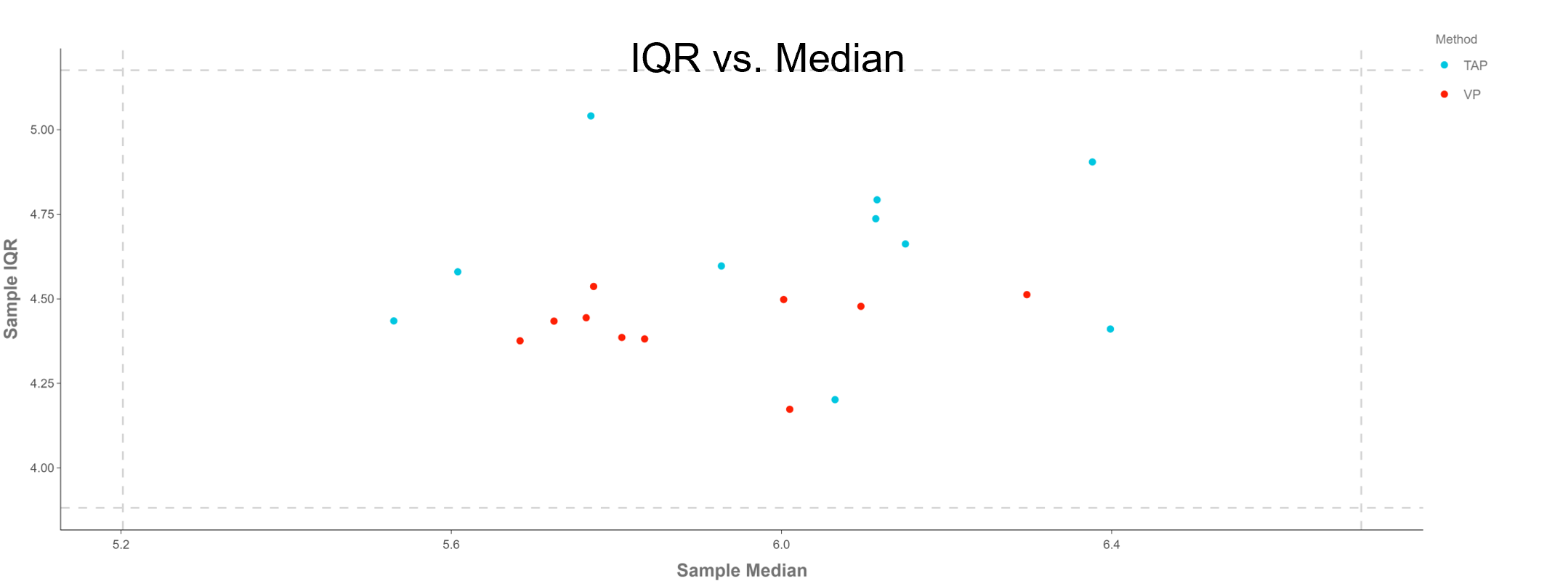
Figure 3: Plot showing the interquartile range (IQR) vs. median. This figure reduces the distribution of NPX values for each sample down to two values; the median and the inter-quartile range (defined as the difference between the 75th and 25th percentiles). Horizontal and vertical dashed lines indicate ±3 standard deviations of all sample medians (x-axis) and IQR’s (y-axis). These are meant only as a guide, not as a criteria for sample exclusion.
Principal component analysis (PCA) was initially conducted to identify any clusters indicative of sample population groupings based on the collection methodology. Components 1 vs 2 were plotted, whereby PCA1 accounts for 33.24% and PCA2 accounts for 16.51% (PCA3 = 9.32%). All 92 proteins were used in the PCA analysis to generate the scatter plot. Donor 7 may be a possible outlier, whereas donors 2 and 9 have QC warnings in the TAP sample. Overall, two distinct groups appear to form between the TAP vs VP samples. 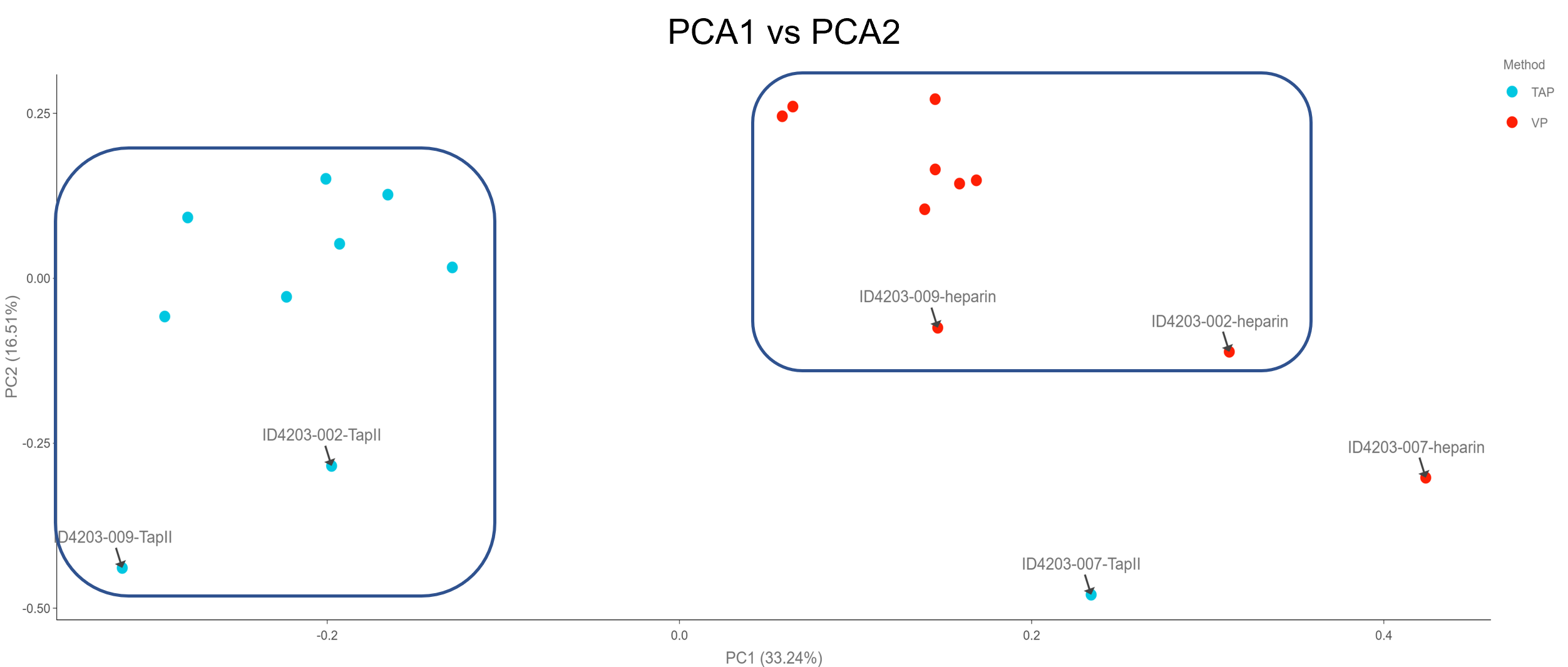
Figure 4: PCA plot showing potential clustering of TAP and VP samples. Potential outliers are highlighted with black arrows.
A heatmap with hierarchical clustering of proteins and samples by collection method was generated to identify any visual patterns. Approximately half of the proteins in the panel appear to be differentially expressed between TAP vs VP samples. The visual groupings are outlined in blue boxes.
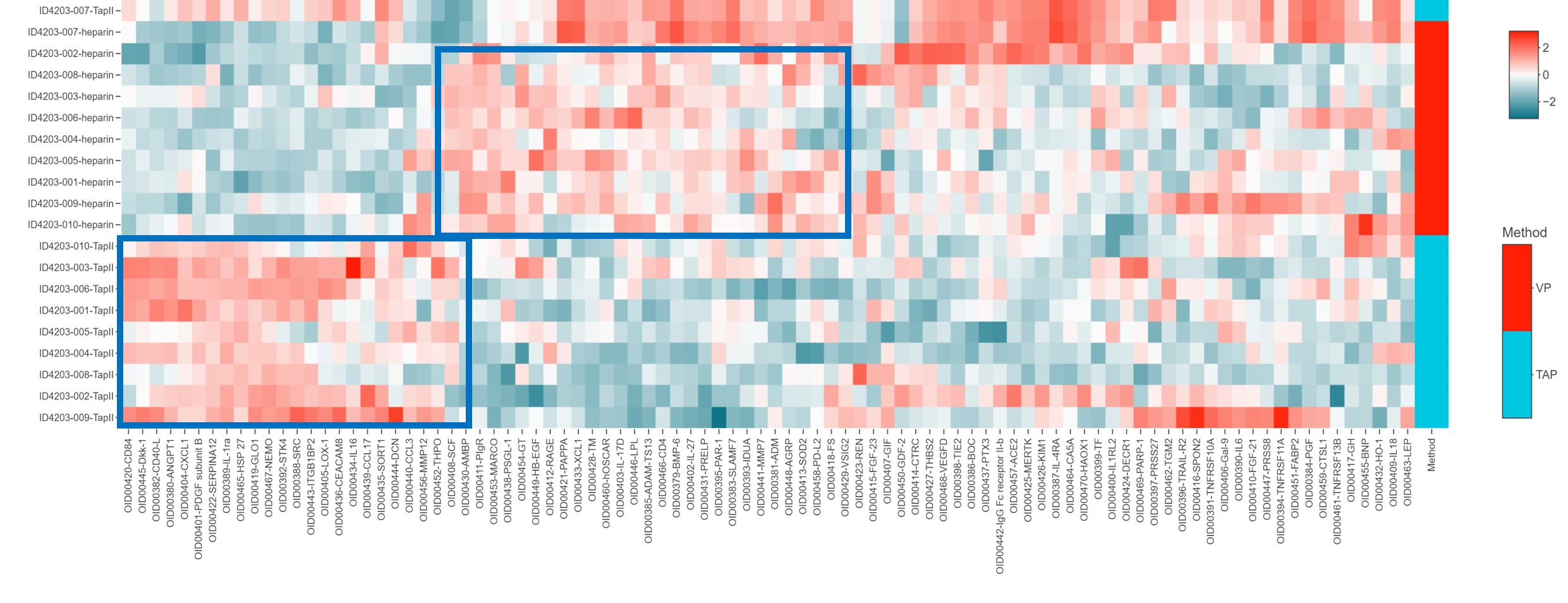
Figure 5: Heat map for visualization of the CVD II panel across all 20 samples. All assay distributions are centered at 0 and scaled to have a standard deviation of 1. Hierarchical clustering based on centered and scaled NPX values is performed on both samples and assays to determine row and column ordering. The centered and scaled NPX values are then used to color the heatmap. Additional method labeling is included.
As an initial protein stratification model to develop two groups, a Welch two sample t-test was performed to understand the statistical significance of detection between the VP and TAP collection methods. Overall, 36 out of 92 proteins have a statistically significant difference in means between the two collection methods. The table shows the full results of the t-test with a volcano plot comparing VP to TAP (with some significant proteins highlighted in the volcano plot). Overall, these statistically-devised groups of proteins were used for further individual donor/analyte analysis (referred to as 36-protein group and 56-protein group).
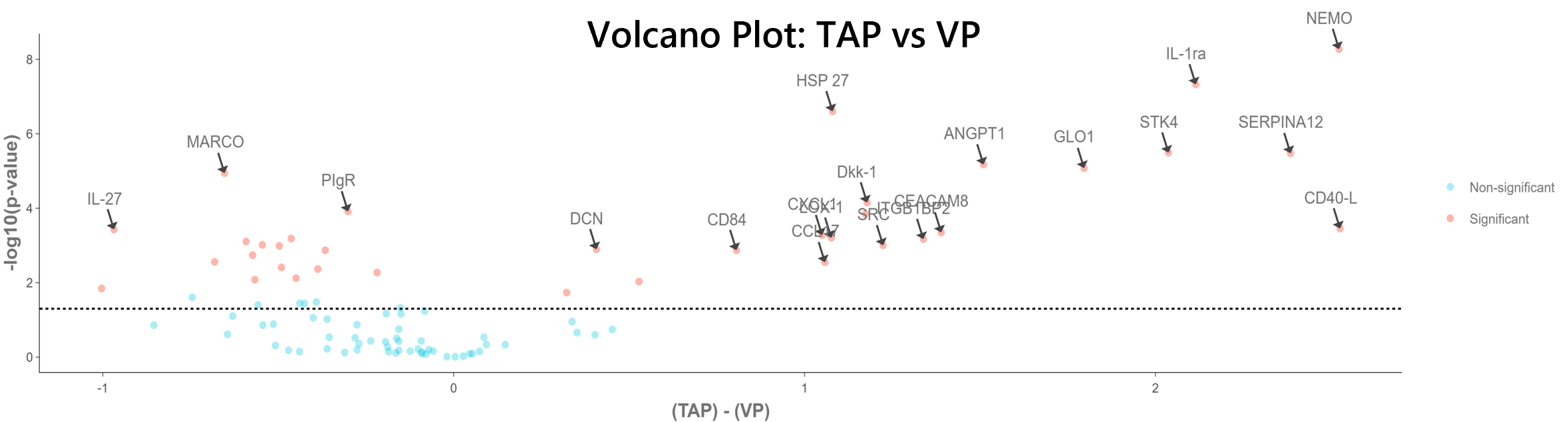
Figure 6: Volcano plot showing significance vs. fold-change as conducted by a t-test. This volcano plot shows the NPX difference between the two groups (TAP vs VP) on the x-axis and the -log10 of the nominal p-value on the y-axis.
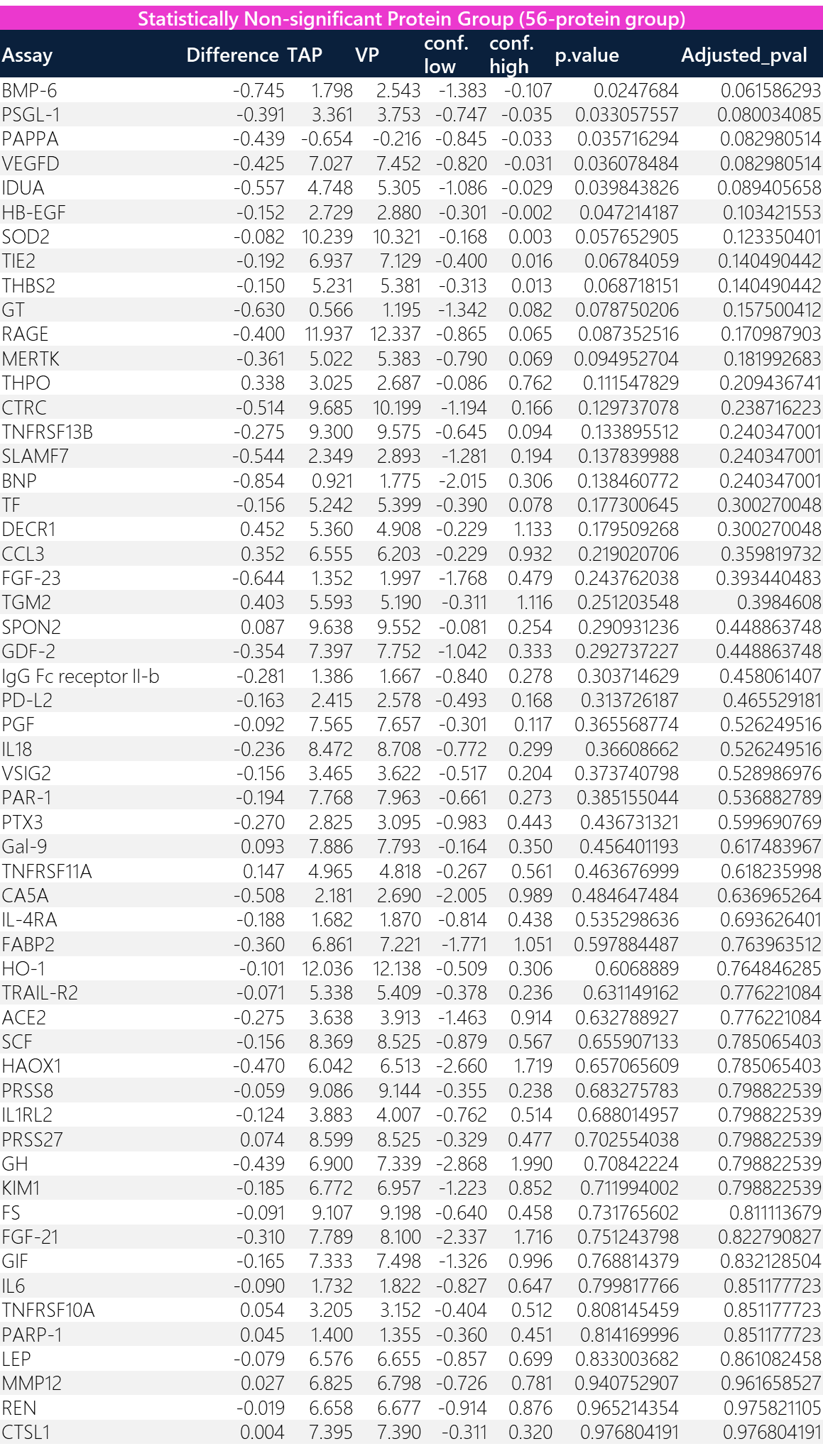
Table 3: Results of the Welch two sample t-test. This hypothetical model stratifies proteins that are of statistical significance (likely different NPX between TAP vs VP) into one group (“36-protein group”) and those that are statistically non-significant (likely the same NPX between TAP vs VP) into another group (“56-protein group”).
To obtain a broad view of trending between TAP and VP, scatter plots were constructed for a high-level assessment of all 92 proteins + 10 donors, the 36-protein group + 10 donors, and the 56-protein group + 10 donors. Bland-Altman plots were constructed to show differences in bias. Overall, there is a greater linear fit for the 56-protein group (R^2 = 0.9852; percentage bias = 4.70%) as compared to the 36-protein group (R^2 = 0.8256; percentage bias = 10.84%).
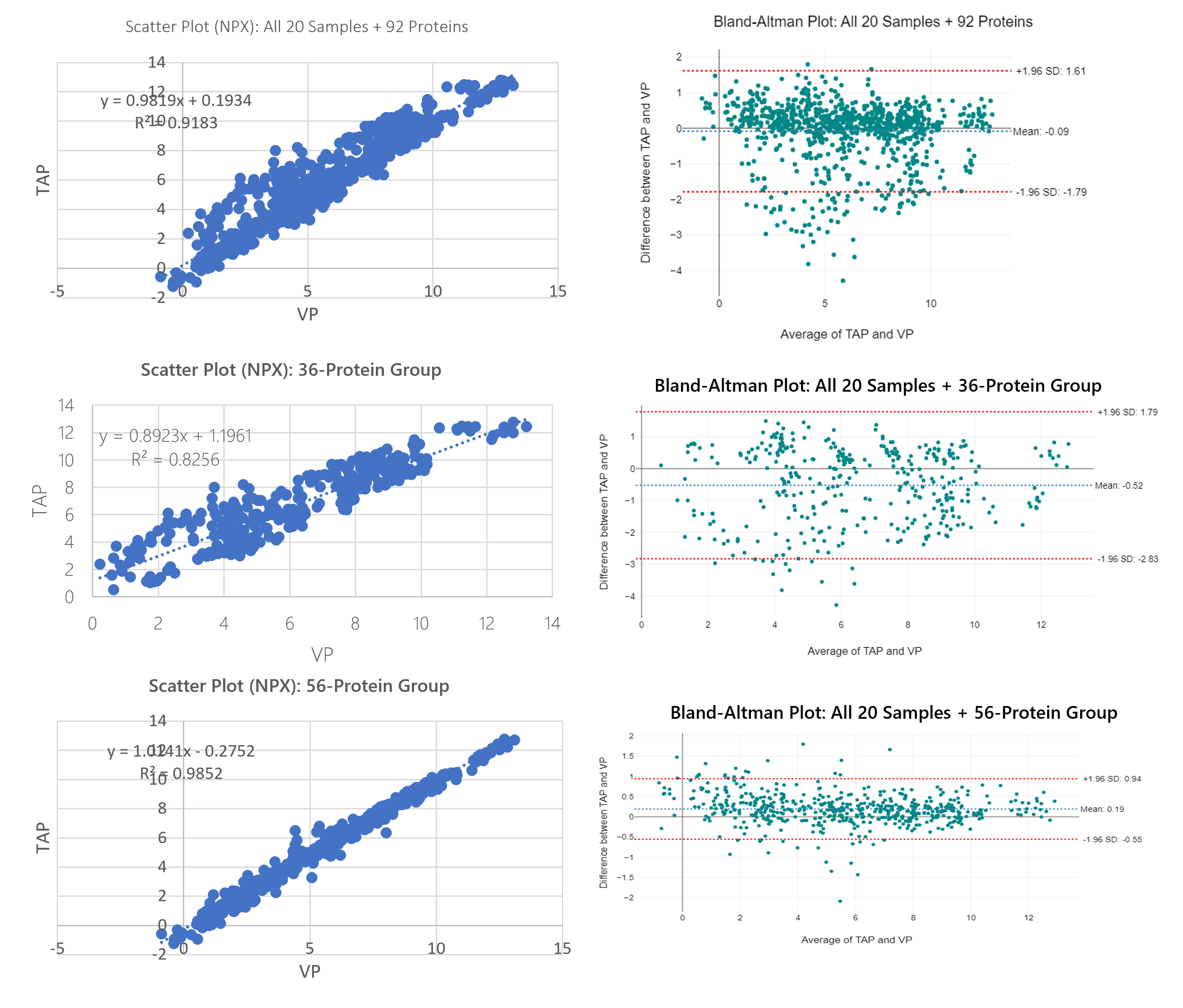
Figure 7: Comprehensive scatter plots for all data vs. 36-protein group vs 56-protein group with respective Bland-Altman plots to broadly highlight the increase in correlation and the reduction in bias between the 36-protein group vs. the 56-protein group. Data is collective of all 10 donors (20 total samples).
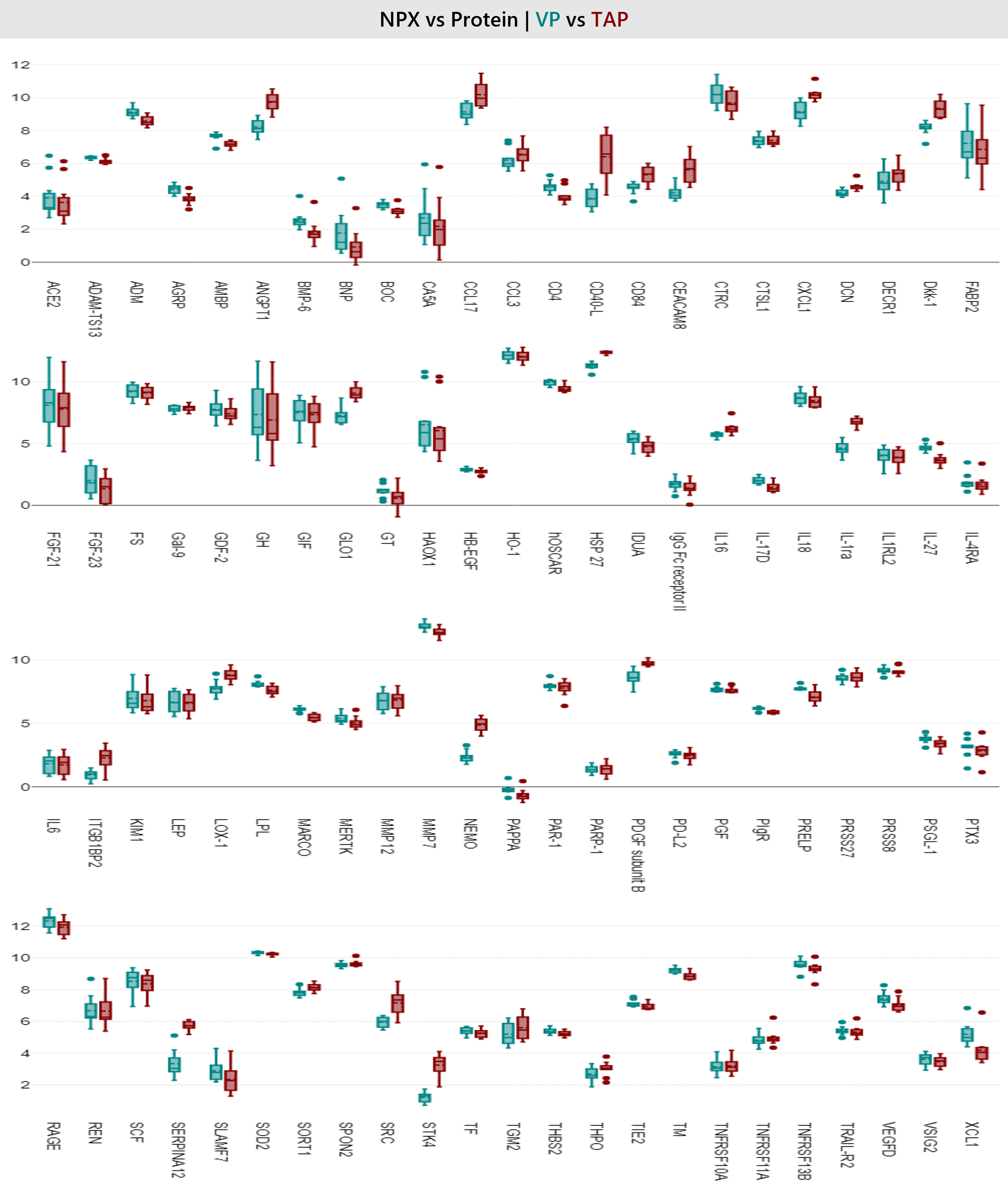
Figure 8: Box plot showing general distribution of individual protein biomarkers for all 92 analytes across all 10 donors between TAP vs VP.
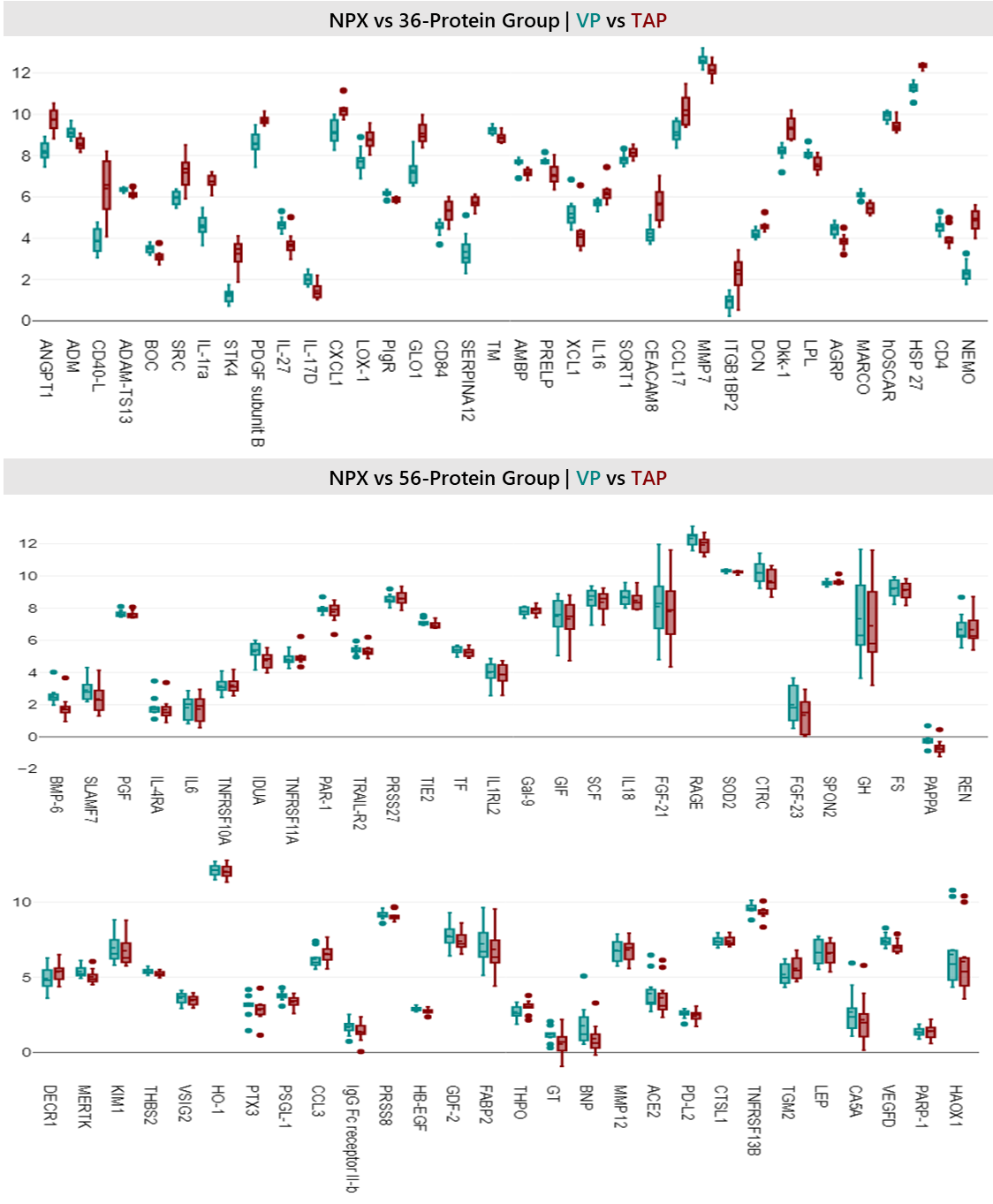
Figure 8: Box plot showing the 36-protein vs 56-protein groups (i.e. statistically significant proteins vs statistically non-significant proteins per t-test stratification) to visually highlight distribution differences between TAP vs VP collection methods across all samples.
A sample correlation assessment was conducted via Pearson and Spearman correlation matrices. As a rudimentary assessment for collective correlation across all samples, the analysis shows good correlation between matched donor pairs of TAP and VP collections. This assessment was based on all 92 proteins.
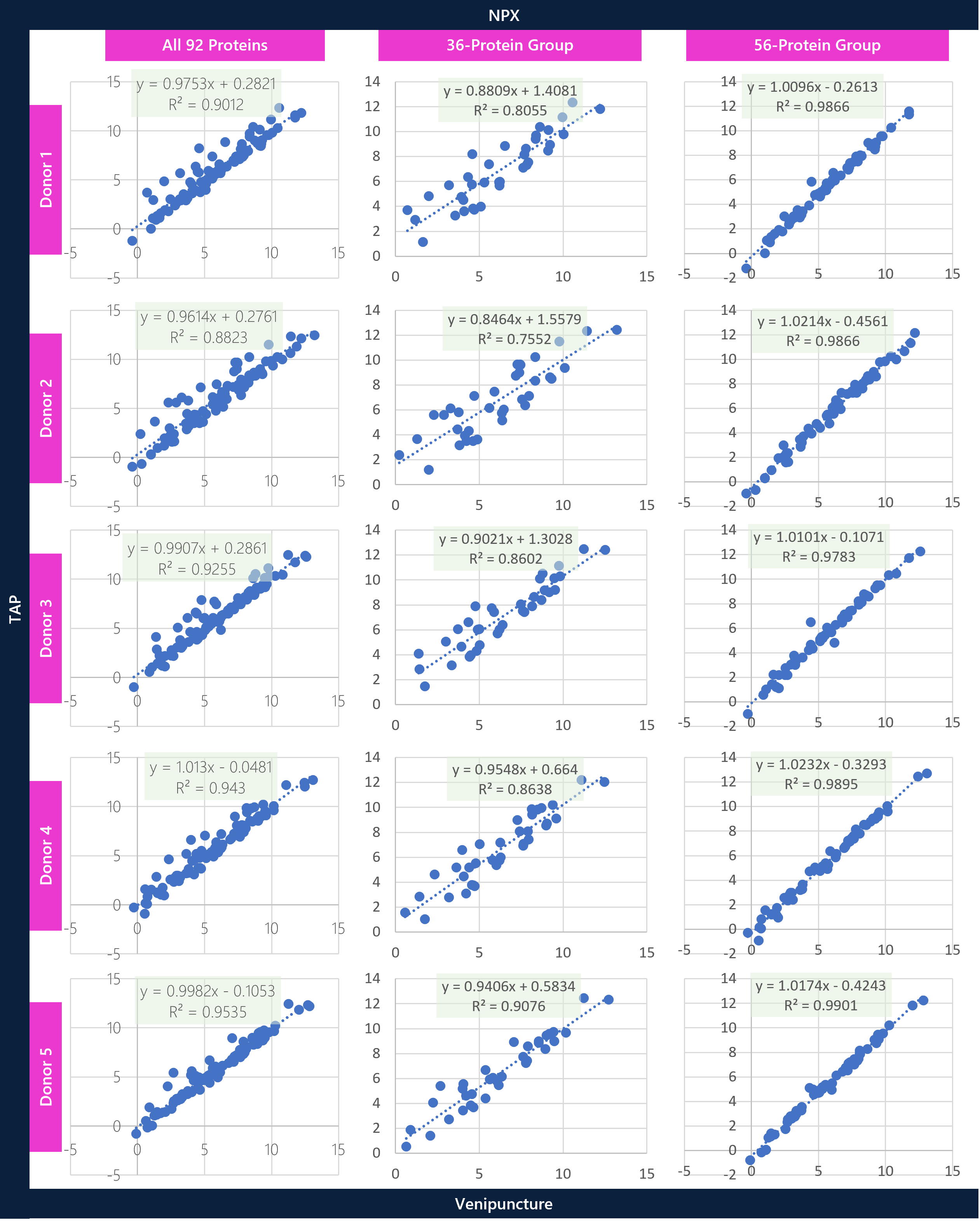
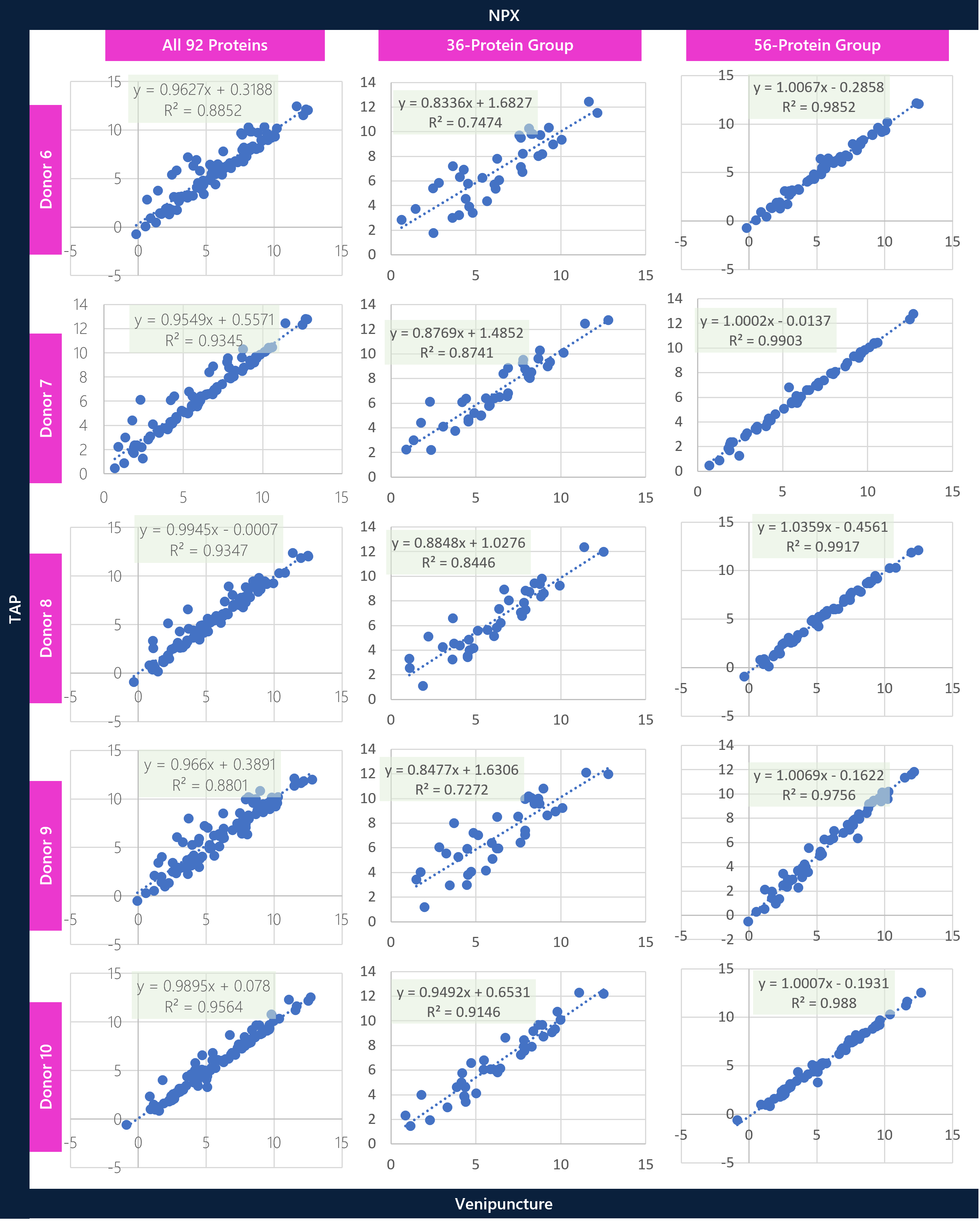
Individual donor assessments between TAP vs VP were conducted to understand the statistically stratified groups of proteins and to better assess concordance across donors. Scatter plots for the full panel (92 proteins), the statistically significant Welch t-test group (36 proteins), and the statistically non-significant Welch t-test group (56 proteins) were prepared to show the differential correlation of proteins between the matrices. Overall, there is good correlation across the entire 92-protein panel, with significantly higher correlation across the stratified 56-protein-group overall. At a broad level, the aim is to show variable differences between matrices for certain proteins prior to analyzing specific proteins in greater detail.
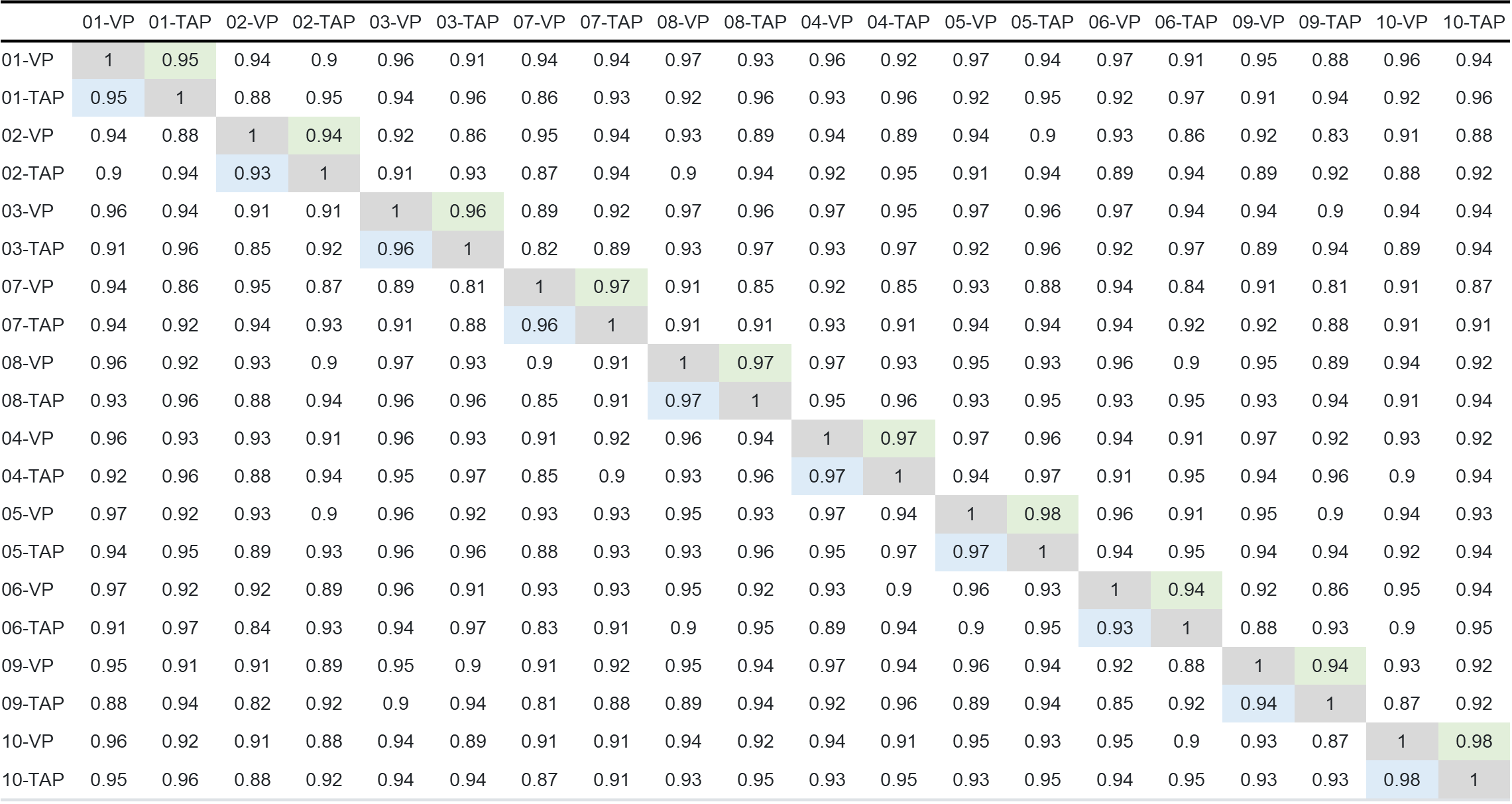
Table 4: Spearman and Pearson correlation matrix across all 20 samples.
CONCLUSIONS
Initial feasibility for Olink Target 96 CVD II panel demonstrates good correlation of NPX values between TAP and VP samples. Specifically, NPX concordance is demonstrated between statistically significant vs. statistically non-significant groups of proteins in addition to the comprehensive totality of proteins within the panel. It is important to highlight that NPX values are a measure of relative expression and not absolute quantitation. Accessible and minimally invasive sampling with the TAP device can enable comprehensive proteomic testing over more frequent timepoints to monitor biological changes. Remote collections with TAP can enable access, throughput and disease monitoring for changes over time when coupled to sensitive and specific PEA technologies such as the Olink platform. The purpose of this study is to demonstrate technology and platform capability of the TAP device with the Olink methodology using the Target 96 kit. Further evaluation of the TAP device could benefit from a larger sample size and comparisons to similar cross-platform technologies to measure the same analytes. Future studies will explore the Olink NGS Explore, whereby absolute quantitation is afforded through “counting” of PEA amplicons via sequencing. Additional considerations may include expansion into other Target 96 panels for similar correlation studies, inclusive of disease vs. non-disease donors for the respective panel (i.e. Target Olink 96 Oncology panel).